



The name SARS-CoV-2 says it directly: Severe Acute Respiratory Syndrome. Yet we continue to see and hear of a stream of new findings that are seemingly inexplicable for a pure respiratory illness: cardiac involvement, hypoxemia out of proportion to ARDS, and severe thrombotic complications. What’s happening? Is COVID-19 a respiratory or a cardiovascular disorder? As clinicians we are guided by insights from pathophysiology and clinical findings through shared experience. So let’s first get our arms around the pathophysiology.
COVID-19 respiratory pathophysiology is well understood: the virus directly attacks the Upper Respiratory Epithelium via ACE2 receptors. After entering these cells and hijacking cellular machinery to replicate, the virus continues to the Lower Respiratory Tract where alveoli may become severely compromised. Susceptible patients present with a flu-like illness, which may progress to pneumonia and ARDS. All of the above happens through initial viral attack on the Respiratory Epithelium along with the ensuing immune response. Indeed patients may present with ‘cytokine storm’, another aspect of the systemic sepsis picture. This sequence is familiar territory for ICU clinicians.
Patients on anticoagulation for a ‘respiratory’ disease?
However, there is a new twist in COVID-19 systemic illness compared with other ‘respiratory’ viral infections. Many other organ systems can become severely affected, with the list growing longer by the day. In addition to insults on specific organs like AKI and myocardial injury, clinicians are reporting a hypercoagulable state. An experienced ICU clinician described to us a thrombus forming and reforming on an internal jugular (IJ) guidewire in the middle of a bedside procedure. And, that COVID-19 patient was already on full anticoagulation! This is not a garden variety hypercoagulable state.
Experienced clinicians are also finding that ‘this is not typical ARDS‘. While classical ARDS occurs in some patients, many COVID-19 patients exhibit severe hypoxemia with normal or near-normal lung mechanics. Increased intrapulmonary shunting as a result of microvascular thrombosis could be the culprit. Indeed, in a previous post, we referred to evidence of Thrombotic Microangiopathy in the lungs (Fox, MedRxiv 2020), not a common finding outside of this disease. Additional pathology reports further support and explain this line of thinking. A biopsy report from five severely ill COVID-19 patients with lung and skin findings showed, “A pattern of tissue damage consistent with complement-mediated microvascular injury,” and “co-localization of SARS-CoV-2-specific spike glycoproteins with complement components in the lung and skin was also documented.”[1] Notably the complement system is primarily an immune cascade yet here participates in microvascular thrombosis. Taking this a step further to the bedside, one very recent clinical case series from Brazil describes a consecutive series of 27 COVID-19 cases treated with full dose heparin.[2]
How do we explain these myriad and seemingly unrelated findings? Viral pneumonia with ARDS and sepsis, disproportionate hypoxemia, intractable thrombosis that is not classical DIC. Nothing ‘typical’ about it. What pathophysiology could possibly tie all of this together?
Look to our cardiovascular system
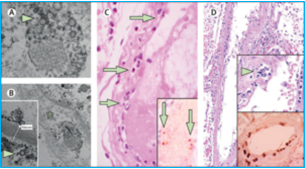
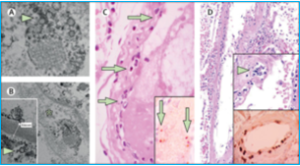 In fact, there is evidence that SARS-CoV-2 directly attacks the vascular endothelium. A small pathology series by Varga et al has definitively shown the presence of viral elements within endothelial cells and an accumulation of inflammatory cells, with evidence of endothelial and inflammatory cell death. Here is direct evidence that COVID-19 patients have “endotheliitis in several organs as a direct consequence of viral involvement and of the host inflammatory response.”[3] What was the result of this endothelial damage? All of the patients in this small series exhibited multiple organ dysfunction and two of the three had documented myocardial dysfunction.
In fact, there is evidence that SARS-CoV-2 directly attacks the vascular endothelium. A small pathology series by Varga et al has definitively shown the presence of viral elements within endothelial cells and an accumulation of inflammatory cells, with evidence of endothelial and inflammatory cell death. Here is direct evidence that COVID-19 patients have “endotheliitis in several organs as a direct consequence of viral involvement and of the host inflammatory response.”[3] What was the result of this endothelial damage? All of the patients in this small series exhibited multiple organ dysfunction and two of the three had documented myocardial dysfunction.
Endothelial involvement is particularly important as this may explain why certain patients are vulnerable. The patients in the Varga series had pre-existing conditions such as hypertension, diabetes, obesity and established cardiovascular disease. These are the same comorbidities reported in many clinical studies of COVID-19 mortality. What do all of these conditions have in common? Endothelial dysfunction.
Vascular endothelium: not just a passive barrier
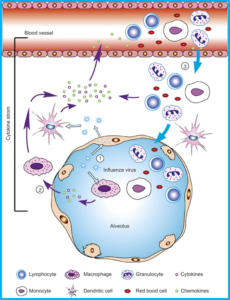 Endothelial cells play an active role in regulating arterial tone. Over the last several years another relevant role has been elucidated: vascular endothelial cells are sentinels of the innate immune system. They are one of the first cells in the body to interact with pathogens, sensing and activating immune cells. Vascular endothelium also plays a significant role in promoting the cytokine storm leading to a variety of downstream events in viral sepsis. In 2011 Tiejaro et al., identified vascular endothelial cells in the lungs as the central orchestrators of the cytokine storm seen in H1N1 disease.[4] Subsequently in 2016, Liu et al., deemed these same endothelial cells “amplifiers” of the cytokine storm in the lung following severe influenza infection.[5]
Endothelial cells play an active role in regulating arterial tone. Over the last several years another relevant role has been elucidated: vascular endothelial cells are sentinels of the innate immune system. They are one of the first cells in the body to interact with pathogens, sensing and activating immune cells. Vascular endothelium also plays a significant role in promoting the cytokine storm leading to a variety of downstream events in viral sepsis. In 2011 Tiejaro et al., identified vascular endothelial cells in the lungs as the central orchestrators of the cytokine storm seen in H1N1 disease.[4] Subsequently in 2016, Liu et al., deemed these same endothelial cells “amplifiers” of the cytokine storm in the lung following severe influenza infection.[5]
The broad range of endothelial function in immunity has been described before: sensing pathogens, activating immune cells, signaling via cytokines, regulating immune activities, and amplifying immune cascades. Now that we have insightful COVID-19 pathophysiology and clinical observations, we can see that SARS-CoV-2 is attacking our two most important barriers simultaneously: respiratory epithelium and vascular endothelium.
COVID-19 endothelial insult paints a mixed clinical picture
Endothelial cells participate in the activation, regulation and amplification of both inflammation and coagulation. These two fundamental homeostatic processes are intimately related, and indeed likely co-evolved. Inflammatory and coagulation pathways both entail complex cascades with the potential for rapid amplification. On the one hand, inflammation can explain pneumonia progressing through ARDS, culminating in sepsis and multiple organ failure. We have seen that endothelial cells exist in the eye of the (cytokine) storm. On the other hand, hyper-coagulation can explain both macro (large vessel) and micro-vascular findings. For example, thrombotic microangiopathy could explain hypoxemia in the face of normal lung mechanics via increased shunting.
So one common underlying mechanism, endothelial damage, can account for two rapidly progressive and damaging states, inflammation and thrombosis. These two states can coexist during one patient’s course, painting a mixed clinical picture. Alternatively, one or the other (inflammation or thrombosis) may be dominant for a given patient. Which process predominates might depend upon the patient population. A common endothelial mechanism likely explains the increased vulnerability of patients with pre-existing conditions such as systemic hypertension and cardiovascular disease.
Perfusion is the key to COVID-19 clinical management
While we await effective therapeutics, our new understanding of COVID-19 as a cardiovascular disorder should guide clinical management of critically ill patients. Given that multiple organ failure precedes COVID-19 mortality, we should focus on perfusion as a basic requirement for organ function. Perfusion is oxygenated blood flow under adequate pressure delivered to each organ system. While maintaining gas exchange is necessary, cardiovascular performance is critical. Since the left ventricle (LV) is responsible for generating pressure and the right ventricle (RV) for maintaining circulatory flow, adequate LV filling and RV function are needed for hemodynamic stability. The ‘last mile’ in delivering flow to the tissues is also crucial, namely microvascular integrity. All three functions must be supported: oxygenation, hemodynamic stability, and microvascular integrity. This is the only way to prevent multiple organ failure and mortality.
For oxygenation, ICU teams are already well versed in using ARDS specific strategies, such as proning, low tidal volume and judicious PEEP. Hemodynamic stability must be addressed with monitoring that affords maintenance of adequate LV filling while avoiding fluid overload. Managing the RV is centrally important, as we already know that the RV is easily overloaded in this condition. Microvascular integrity may be supported by anticoagulation strategies, which are evolving as new information develops.
COVID-19 is a new systemic disease with both respiratory and cardiovascular components. For the most critically ill, focusing on organ perfusion can give our patients the time needed to survive the initial onslaught. Support oxygenation, stabilize hemodynamics, and promote microvascular integrity.
Now is the time to upgrade our clinical capabilities. Let’s talk about how.
[1] Magro C, Mulvey JJ, Berlin D, et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: A report of five cases [published online ahead of print, 2020 Apr 15]. Transl Res. 2020;S1931-5244(20)30070-0. doi:10.1016/j.trsl.2020.04.007
[2] Negri EM, Piloto B, Morinaga LK, et al. Heparin therapy improving hypoxia in COVID-19 patients – a case series. MedRxiv 2020.04.15.20067017. doi.org/10.1101/2020.04.15.20067017
[3] Varga Z, Flammer A, Steiger P, et al. Endothelial cell infection and endotheliitis in COVID-19. [published online 2020 Apr 17]. Lancet. doi.org/10.1016/ S0140-6736(20)30937-5
[4] Teijaro JR, Walsh KB, Cahalan S, et al. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011. 146: 980–991. doi:10.1016/j.cell.2011.08.015
[5] Liu Q, Zhou Y, Yang Z. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell Mol Immunol. 2016. 13:3–10. doi.org/10.1038/cmi.2015.74